Differential testing framework
STEP13 Differential gene testing
Differential gene testing is a critical component of single-cell analysis. In the past people have used single-cell testing algorithms to look at differentially expressed genes, however, newer computational studies argue that pseduobulking counts across cell types and donors and then performing gene testing is perhaps more appropriate when discovering differentially expressed genes. To read more about this, review this paper by Squair et al., 2021:Nature Communications. However based on the design of the experiment and the cutoffs, both can yield reproducible data, at least for highly differential genes, for genes with high variance, perhaps pseudobulk methods are more reliable.
STEP13A: Single-cell gene testing
# In order to fine DE genes its important to annotate cells,
# this is where metadata comes to be important
# Our first method employs single cell differential gene expression using LR
# First lets save celltype info from active.idents into a metadata slot called celltype
pbmc$celltype <- pbmc@active.ident
# Lets check the clusters
table(pbmc$celltype)
# Now lets make a new metadata slot containing both peices of information, celltype and stimulation with interferon
# In order to do this we need to take data from two slots 1) pbmc$celltype and 2) pbmc$stim
# We will store this data in a new metadata slot called celltype.stim
pbmc$celltype.stim <- paste(pbmc$celltype, pbmc$stim, sep = "_")
# Now lets load this metadata in the active directory of seurat, so that seurat will pull this metadata by default
Idents(pbmc) <- "celltype.stim"
# Now lets run differential analysis
# HINT: Note that the ident.1 and ident.2 terms reside in pbmc$celltype.stim. maybe run table(pbmc$celltype.stim) to see a list of terms?
# If you cant complete all the steps to get here, thats okay! I have a seurat object called PBMC saved in a shared folder in a sub-folder called: \seuratobjects
# Just download that object and use qread or readRDS to load your object into R
b.interferon.response <- FindMarkers(pbmc, ident.1 = "B_STIM", ident.2 = "B_CTRL", # These are the two cell types/clusters that you want to run a DE test across. ident.2 is your control
slot = 'data',
test.use = "LR",
min.pct = 0.1,
latent.vars = c("donor"),
logfc.threshold = 0.5849, #~1.5FC
only.pos = TRUE,
verbose = FALSE)
# Got the DE test to run for your project? WOOHOO GREAT JOB!!! I knew you could do it!!
# If you couldn't DON'T WORRY, create an issue on GitHub and I will get back to you asap!!
# Output
Warning message:
glm.fit: fitted probabilities numerically 0 or 1 occurred
# Lets check head of data
head(b.interferon.response, n = 15)
We are now looking at the output of differentially expressed genes using the single cell statistical method
#OUTPUT
p_val avg_log2FC pct.1 pct.2 p_val_adj
ISG15 1.886870e-251 4.5777825 0.998 0.244 2.651618e-247
ISG20 6.775871e-233 2.9286888 1.000 0.674 9.522132e-229
IFIT3 2.249518e-226 4.5012181 0.963 0.052 3.161247e-222
IFI6 7.675338e-222 4.2569268 0.965 0.077 1.078615e-217
IFIT1 1.235784e-197 4.1209722 0.908 0.032 1.736647e-193
MX1 1.447618e-160 3.2484413 0.908 0.116 2.034337e-156
TNFSF10 1.295544e-148 3.7724262 0.781 0.022 1.820627e-144
LY6E 1.309127e-148 3.1014142 0.894 0.151 1.839716e-144
IFIT2 4.554090e-142 3.6335959 0.786 0.037 6.399863e-138
B2M 2.530957e-119 0.6158652 1.000 1.000 3.556754e-115
CXCL10 4.799024e-113 5.3217240 0.644 0.010 6.744069e-109
PLSCR1 2.659608e-112 2.8015323 0.788 0.119 3.737548e-108
IRF7 3.667981e-108 2.5623663 0.837 0.193 5.154613e-104
HERC5 6.244788e-98 2.8136999 0.611 0.022 8.775800e-94
UBE2L6 7.032047e-91 2.1202128 0.857 0.301 9.882136e-87
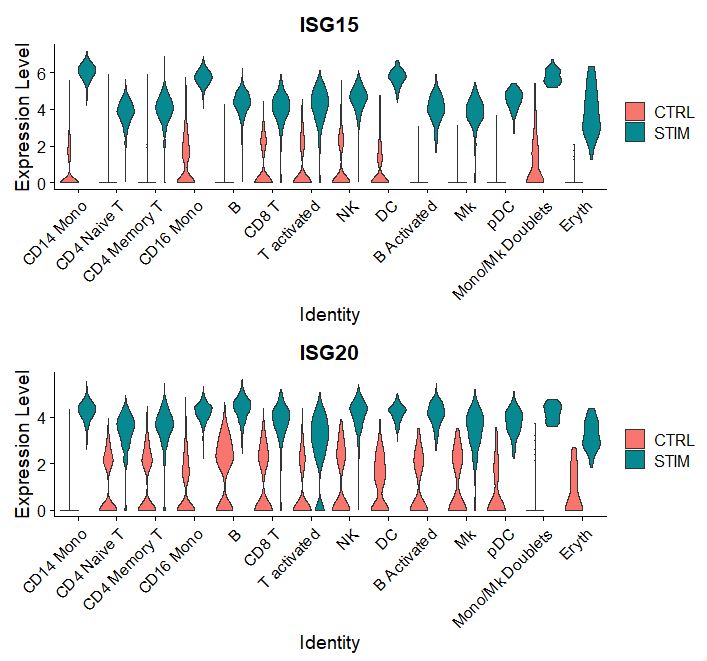
Let’s look at the gene expression of the top 2 candidate proteins, we can see that they are interferon response genes.
b.interferon.response <- dplyr::filter(b.interferon.response, p_val_adj < 5e-2)
plots <- VlnPlot(pbmc, features = c("ISG15", "ISG20"), split.by = "stim", group.by = "celltype",
pt.size = 0, combine = FALSE)
#OUTPUT
The default behaviour of split.by has changed.
Separate violin plots are now plotted side-by-side.
To restore the old behaviour of a single split violin,
set split.plot = TRUE.
This message will be shown once per session.
# Run plotting function
wrap_plots(plots = plots, ncol = 1)
#OUTPUT
STEP13B: Pseudobulk testing
Pseudobulking is powerful. It allows one to remove the influence of library composition bias, as one aggregated read now represents the expression of one celltype across different donors. This analytical philosophy albeit more conservative, yields superior results with regards to reproducibility.
DefaultAssay(pbmc) <- "RNA"
pbmc$celltype.stim.donor <- paste(pbmc$celltype, pbmc$stim, pbmc$donor, sep = "_")
Idents(pbmc) <- "celltype.stim.donor"
table(pbmc[["celltype.stim.donor"]])
celltype.stim.donor
#OUTPUT
B Activated_CTRL_d1 B Activated_CTRL_d2 B Activated_CTRL_d3 B Activated_CTRL_d4
41 46 50 46
B Activated_STIM_d1 B Activated_STIM_d2 B Activated_STIM_d3 B Activated_STIM_d4
50 66 44 47
B_CTRL_d1 B_CTRL_d2 B_CTRL_d3 B_CTRL_d4
110 109 93 93
B_STIM_d1 B_STIM_d2 B_STIM_d3 B_STIM_d4
136 141 145 143
CD14 Mono_CTRL_d1 CD14 Mono_CTRL_d2 CD14 Mono_CTRL_d3 CD14 Mono_CTRL_d4
534 582 520 573
CD14 Mono_STIM_d1 CD14 Mono_STIM_d2 CD14 Mono_STIM_d3 CD14 Mono_STIM_d4
551 535 504 524
CD16 Mono_CTRL_d1 CD16 Mono_CTRL_d2 CD16 Mono_CTRL_d3 CD16 Mono_CTRL_d4
125 124 126 143
CD16 Mono_STIM_d1 CD16 Mono_STIM_d2 CD16 Mono_STIM_d3 CD16 Mono_STIM_d4
142 141 127 136
CD4 Memory T_CTRL_d1 CD4 Memory T_CTRL_d2 CD4 Memory T_CTRL_d3 CD4 Memory T_CTRL_d4
219 200 208 186
CD4 Memory T_STIM_d1 CD4 Memory T_STIM_d2 CD4 Memory T_STIM_d3 CD4 Memory T_STIM_d4
229 197 238 239
CD4 Naive T_CTRL_d1 CD4 Naive T_CTRL_d2 CD4 Naive T_CTRL_d3 CD4 Naive T_CTRL_d4
264 230 253 256
CD4 Naive T_STIM_d1 CD4 Naive T_STIM_d2 CD4 Naive T_STIM_d3 CD4 Naive T_STIM_d4
379 367 346 383
CD8 T_CTRL_d1 CD8 T_CTRL_d2 CD8 T_CTRL_d3 CD8 T_CTRL_d4
71 72 82 95
CD8 T_STIM_d1 CD8 T_STIM_d2 CD8 T_STIM_d3 CD8 T_STIM_d4
129 119 101 117
DC_CTRL_d1 DC_CTRL_d2 DC_CTRL_d3 DC_CTRL_d4
65 61 52 48
DC_STIM_d1 DC_STIM_d2 DC_STIM_d3 DC_STIM_d4
48 39 50 57
Eryth_CTRL_d1 Eryth_CTRL_d2 Eryth_CTRL_d3 Eryth_CTRL_d4
5 5 6 6
Eryth_STIM_d1 Eryth_STIM_d2 Eryth_STIM_d3 Eryth_STIM_d4
7 9 10 7
Mk_CTRL_d1 Mk_CTRL_d2 Mk_CTRL_d3 Mk_CTRL_d4
21 30 26 21
Mk_STIM_d1 Mk_STIM_d2 Mk_STIM_d3 Mk_STIM_d4
29 28 37 28
Mono/Mk Doublets_CTRL_d1 Mono/Mk Doublets_CTRL_d2 Mono/Mk Doublets_CTRL_d3 Mono/Mk Doublets_CTRL_d4
17 5 9 11
Mono/Mk Doublets_STIM_d1 Mono/Mk Doublets_STIM_d2 Mono/Mk Doublets_STIM_d3 Mono/Mk Doublets_STIM_d4
4 7 11 6
NK_CTRL_d1 NK_CTRL_d2 NK_CTRL_d3 NK_CTRL_d4
82 63 77 90
NK_STIM_d1 NK_STIM_d2 NK_STIM_d3 NK_STIM_d4
75 85 86 87
pDC_CTRL_d1 pDC_CTRL_d2 pDC_CTRL_d3 pDC_CTRL_d4
10 15 9 17
pDC_STIM_d1 pDC_STIM_d2 pDC_STIM_d3 pDC_STIM_d4
13 23 16 25
T activated_CTRL_d1 T activated_CTRL_d2 T activated_CTRL_d3 T activated_CTRL_d4
83 79 84 69
T activated_STIM_d1 T activated_STIM_d2 T activated_STIM_d3 T activated_STIM_d4
92 91 83 77
combined_pbmc <- AggregateExpression(pbmc,
assays = c("RNA"),
features = NULL, return.seurat = TRUE,
group.by = "celltype.stim.donor",
slot = "counts", verbose = FALSE)
combined_pbmc$celltype.stim.donor <- Cells(combined_pbmc)
# Metadata organization and addition to aggregated object
{
Idents(combined_pbmc) <- 'celltype.stim.donor'
combined_pbmc$celltype <- combined_pbmc@meta.data[["orig.ident"]]
metadat <- combined_pbmc@meta.data
metadat$celltype <- metadat[c('celltype')] <- str_split_i(metadat$celltype.stim.donor, "_", -3)
metadat$stim <- metadat[c('stim')] <- str_split_i(metadat$celltype.stim.donor, '_', -2)
metadat$donor <- metadat[c('donor')] <- str_split_i(metadat$celltype.stim.donor, '_', -1)
combined_pbmc@meta.data = metadat
}
table(combined_pbmc@meta.data[["celltype"]])
#OUTPUT
B B Activated CD14 Mono CD16 Mono CD4 Memory T CD4 Naive T
8 8 8 8 8 8
CD8 T DC Eryth Mk Mono/Mk Doublets NK
8 8 8 8 8 8
pDC T activated
8 8
table(combined_pbmc@meta.data[["donor"]])
#OUTPUT
d1 d2 d3 d4
28 28 28 28
table(combined_pbmc@meta.data[["celltype.stim.donor"]])
#OUTPUT
B Activated_CTRL_d1 B Activated_CTRL_d2 B Activated_CTRL_d3 B Activated_CTRL_d4
1 1 1 1
B Activated_STIM_d1 B Activated_STIM_d2 B Activated_STIM_d3 B Activated_STIM_d4
1 1 1 1
B_CTRL_d1 B_CTRL_d2 B_CTRL_d3 B_CTRL_d4
1 1 1 1
B_STIM_d1 B_STIM_d2 B_STIM_d3 B_STIM_d4
1 1 1 1
CD14 Mono_CTRL_d1 CD14 Mono_CTRL_d2 CD14 Mono_CTRL_d3 CD14 Mono_CTRL_d4
1 1 1 1
CD14 Mono_STIM_d1 CD14 Mono_STIM_d2 CD14 Mono_STIM_d3 CD14 Mono_STIM_d4
1 1 1 1
CD16 Mono_CTRL_d1 CD16 Mono_CTRL_d2 CD16 Mono_CTRL_d3 CD16 Mono_CTRL_d4
1 1 1 1
CD16 Mono_STIM_d1 CD16 Mono_STIM_d2 CD16 Mono_STIM_d3 CD16 Mono_STIM_d4
1 1 1 1
CD4 Memory T_CTRL_d1 CD4 Memory T_CTRL_d2 CD4 Memory T_CTRL_d3 CD4 Memory T_CTRL_d4
1 1 1 1
CD4 Memory T_STIM_d1 CD4 Memory T_STIM_d2 CD4 Memory T_STIM_d3 CD4 Memory T_STIM_d4
1 1 1 1
CD4 Naive T_CTRL_d1 CD4 Naive T_CTRL_d2 CD4 Naive T_CTRL_d3 CD4 Naive T_CTRL_d4
1 1 1 1
CD4 Naive T_STIM_d1 CD4 Naive T_STIM_d2 CD4 Naive T_STIM_d3 CD4 Naive T_STIM_d4
1 1 1 1
CD8 T_CTRL_d1 CD8 T_CTRL_d2 CD8 T_CTRL_d3 CD8 T_CTRL_d4
1 1 1 1
CD8 T_STIM_d1 CD8 T_STIM_d2 CD8 T_STIM_d3 CD8 T_STIM_d4
1 1 1 1
DC_CTRL_d1 DC_CTRL_d2 DC_CTRL_d3 DC_CTRL_d4
1 1 1 1
DC_STIM_d1 DC_STIM_d2 DC_STIM_d3 DC_STIM_d4
1 1 1 1
Eryth_CTRL_d1 Eryth_CTRL_d2 Eryth_CTRL_d3 Eryth_CTRL_d4
1 1 1 1
Eryth_STIM_d1 Eryth_STIM_d2 Eryth_STIM_d3 Eryth_STIM_d4
1 1 1 1
Mk_CTRL_d1 Mk_CTRL_d2 Mk_CTRL_d3 Mk_CTRL_d4
1 1 1 1
Mk_STIM_d1 Mk_STIM_d2 Mk_STIM_d3 Mk_STIM_d4
1 1 1 1
Mono/Mk Doublets_CTRL_d1 Mono/Mk Doublets_CTRL_d2 Mono/Mk Doublets_CTRL_d3 Mono/Mk Doublets_CTRL_d4
1 1 1 1
Mono/Mk Doublets_STIM_d1 Mono/Mk Doublets_STIM_d2 Mono/Mk Doublets_STIM_d3 Mono/Mk Doublets_STIM_d4
1 1 1 1
NK_CTRL_d1 NK_CTRL_d2 NK_CTRL_d3 NK_CTRL_d4
1 1 1 1
NK_STIM_d1 NK_STIM_d2 NK_STIM_d3 NK_STIM_d4
1 1 1 1
pDC_CTRL_d1 pDC_CTRL_d2 pDC_CTRL_d3 pDC_CTRL_d4
1 1 1 1
pDC_STIM_d1 pDC_STIM_d2 pDC_STIM_d3 pDC_STIM_d4
1 1 1 1
T activated_CTRL_d1 T activated_CTRL_d2 T activated_CTRL_d3 T activated_CTRL_d4
1 1 1 1
T activated_STIM_d1 T activated_STIM_d2 T activated_STIM_d3 T activated_STIM_d4
1 1 1 1
combined_pbmc$celltype.stim <- paste(combined_pbmc$celltype, combined_pbmc$stim, sep = "_")
table(combined_pbmc@meta.data[["celltype.stim"]])
#OUTPUT
B Activated_CTRL B Activated_STIM B_CTRL B_STIM CD14 Mono_CTRL
4 4 4 4 4
CD14 Mono_STIM CD16 Mono_CTRL CD16 Mono_STIM CD4 Memory T_CTRL CD4 Memory T_STIM
4 4 4 4 4
CD4 Naive T_CTRL CD4 Naive T_STIM CD8 T_CTRL CD8 T_STIM DC_CTRL
4 4 4 4 4
DC_STIM Eryth_CTRL Eryth_STIM Mk_CTRL Mk_STIM
4 4 4 4 4
Mono/Mk Doublets_CTRL Mono/Mk Doublets_STIM NK_CTRL NK_STIM pDC_CTRL
4 4 4 4 4
pDC_STIM T activated_CTRL T activated_STIM
4 4 4
Idents(combined_pbmc) <- "celltype.stim"
b.interferon.response.aggr <- FindMarkers(combined_pbmc, ident.1 = "B_STIM", ident.2 = "B_CTRL",
slot = 'data',
test.use = "DESeq2",
min.pct = 0.1,
latent.vars = c("donor"),
logfc.threshold = 0.5849, #~1.5FC
only.pos = TRUE,
verbose = FALSE)
Warning: 'latent.vars' is only used for the following tests: negbinom, poisson, MAST, LR
converting counts to integer mode
gene-wise dispersion estimates
mean-dispersion relationship
final dispersion estimates
head(b.interferon.response.aggr, n = 15)
#OUTPUT
p_val avg_log2FC pct.1 pct.2 p_val_adj
ISG15 0.000000e+00 5.458484 1 1.0 0.000000e+00
ISG20 0.000000e+00 3.548164 1 1.0 0.000000e+00
IFI6 1.318852e-111 5.919998 1 1.0 1.853383e-107
IFIT3 1.038933e-98 6.333155 1 1.0 1.460013e-94
LY6E 6.543563e-98 4.196682 1 1.0 9.195669e-94
MX1 1.404512e-97 4.453682 1 1.0 1.973761e-93
B2M 1.505428e-79 1.096096 1 1.0 2.115578e-75
IFIT1 7.894356e-69 6.282988 1 1.0 1.109394e-64
IRF7 3.917788e-68 3.564378 1 1.0 5.505668e-64
CXCL10 6.061481e-61 8.071050 1 0.5 8.518199e-57
IFIT2 6.308882e-61 5.546609 1 1.0 8.865872e-57
PLSCR1 2.569268e-60 4.125013 1 1.0 3.610593e-56
UBE2L6 8.745682e-60 2.935460 1 1.0 1.229031e-55
IFITM2 1.077820e-55 3.460921 1 1.0 1.514660e-51
SAT1 1.858432e-55 2.519988 1 1.0 2.611655e-51
Notice how some adj pvalues are 0? Thats because the value is lower than the smallest calculable value of R, we can see what is the smallest value R can calculate using the following function. Remember this will change based on the hardware you are using.
# Why is my adj pval 0?
.Machine$double.xmin
#OUTPUT
[1] 2.225074e-308
# Replace 0 with low pval, subset for 0.05 FDR and FC > 1.5
b.interferon.response.aggr$p_val_adj[b.interferon.response.aggr$p_val_adj == 0] <- 2e-302
b.interferon.response.aggr <- dplyr::filter(b.interferon.response.aggr, p_val_adj < 5e-2)
b.interferon.response.aggr <- dplyr::filter(b.interferon.response.aggr, avg_log2FC > 0.5849)
intersect(rownames(b.interferon.response), rownames(b.interferon.response.aggr))
#OUTPUT
[1] "ISG15" "ISG20" "IFIT3" "IFI6" "IFIT1" "MX1" "TNFSF10" "LY6E" "IFIT2"
[10] "B2M" "CXCL10" "PLSCR1" "IRF7" "HERC5" "UBE2L6" "IFI44L" "EPSTI1" "OAS1"
[19] "GBP1" "IFITM2" "SAMD9L" "NT5C3A" "IFI35" "PSMB9" "MX2" "DYNLT1" "BST2"
[28] "IFITM3" "CMPK2" "SAT1" "EIF2AK2" "PPM1K" "GBP4" "DDX58" "PSMA2.1" "LAP3"
[37] "SAMD9" "XAF1" "IFI16" "COX5A" "SOCS1" "MYL12A" "SP110" "PARP14" "PSME2"
[46] "TMSB10" "CHST12" "FBXO6" "MT2A" "PLAC8" "TRIM22" "DRAP1" "SUB1" "TNFSF13B"
[55] "NMI" "XRN1" "NEXN" "RBCK1" "CLEC2D" "MNDA" "RNF213" "IFI44" "GBP5"
[64] "NPC2" "STAT1" "WARS" "OAS2" "SELL" "TAP1" "DDX60L" "IRF8" "OAS3"
[73] "RTCB" "IFITM1" "KIAA0040" "CXCL11" "CARD16" "PSMA4" "DNAJA1" "IFIH1" "TYMP"
[82] "HLA-E" "LGALS9" "NUB1" "C19orf66" "GBP2" "PSMB8" "GNG5" "HAPLN3" "PMAIP1"
[91] "IFIT5" "PARP9" "CD38" "GMPR" "C5orf56" "EAF2" "HERC6" "CD48" "RTP4"
[100] "RABGAP1L" "USP30-AS1" "TREX1" "IGFBP4" "INPP1" "CCL8" "CREM" "CD164" "CLIC1"
[109] "APOL6" "CCL2" "SMCHD1" "ODF2L" "EHD4" "NAPA" "SP100" "PHF11" "FAM46A"
[118] "PNPT1" "ADAR" "POMP" "UNC93B1" "DCK" "IRF1" "CCR7" "TMEM123" "CASP4"
[127] "HSH2D" "SLFN5" "CFLAR" "CASP1" "VAMP5" "PSME1" "XBP1" "MRPL44"