Introduction
An astounding degree of diversity exists across human tissue. In order to evaluate cellular heterogeneity across cell types, scRNAseq offers a method to understand underlying cellular gene expression patterns. These expression patters which define cellular states can be used to infer cell function in non-diseases and diseased cell types. We can use scRNAseq to define cell types in a tissue, characterize rare cells, visualize changes over time occuring in the context of development of drug therapy, identify rare low epressed genes, incorporate multiomic data with regards to accessible genome and protein expression data.
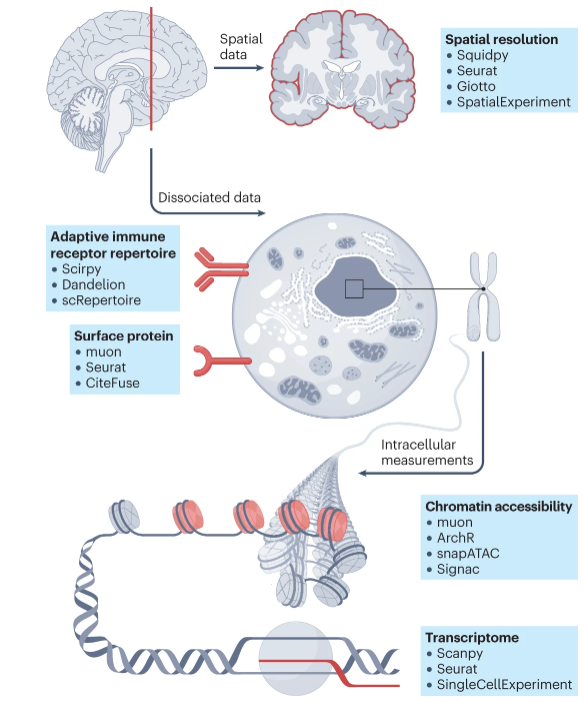
Figure adaped from Heumos et al., 2023: Nature Reviews Genetics
Challenges of scRNAseq analysis
Before the advent of scRNAseq we would perform bulk RNAseq, where we would take a group of cells and isolate RNA convert that RNA into a cDNA library and then subject to sanger sequencing. This is still a great way to analyze average tissue expression of a gene, and owing to higher depth of sequencing, can be used to study genes with low RNA expression. This is a great methos if you are not concerned about cellular heterogeneity and are looking to see the effect of a treatment/drug or overall average effect on a group of cells. While this remains to be a powerful technique, it still does not allow you to study cellular heterogeneity across tissue types or cellular states. For example when looking at gene expression for a certain set of genes we can be mislead by divergent effects of two seperate groups of cells in a collectively sequenced bulk tissue sample.
The biggest challenge with scRNAseq data is cost and the number of datapoints. In order to analyze and process this data an incredible amount of computing power and computing architecture is required to appropriately analyze and interpret scRNAseq data. It is for this reason that scRNAseq analysis remains a challenge for many biologists today. The major challenges for scRNAseq data are large data volume, low sequencing depth/cell, biological variability, technical variability and computational power. The accuracy and sensitivity of bulk/batch experiments remains the highest, so its important to ask yourself if you truly need to perform scRNAseq with regards to your experiment or can you just perform bulk-RNAseq and get more accurate and sensitive results?
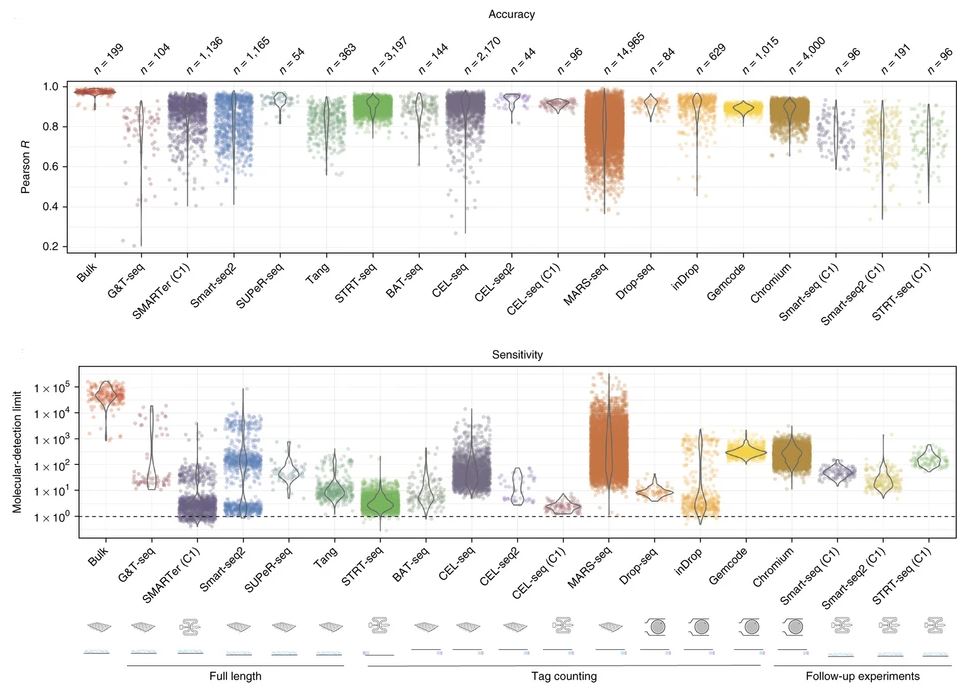 Figure adapted from: Svensson et al., 2017: Nature Methods
Figure adapted from: Svensson et al., 2017: Nature Methods